Our Research
How does altered morphogenesis of the cranial mesenchyme result in neural tube defects?
Neural tube defects are among the most common structural birth defects in humans and result in long-term disability or even death; yet, the underlying genetic causes remain largely unknown. Work in the Zohn Laboratory is guided by the idea that addressing this knowledge gap is best achieved by elucidating the mechanisms mediating both normal and abnormal development. Neural tube defects result from failure of the flat neural plate — the embryonic precursor to the central nervous system — to roll into a tube. In the head, morphogenesis requires movement of not only the cells in the neural plate but also the underlying cranial mesenchyme. Experiments conducted nearly 40 years ago suggest that expansion of the cranial mesenchyme, a cell population beneath the neural plate, is required for elevation of the cranial neural folds and neural tube closure. Yet, little is known regarding how expansion of the cranial mesenchyme drives neural fold elevation and how this process can be disrupted to cause neural tube defects. Moreover, few genes are implicated. To address these gaps in knowledge, we are utilizing a novel model of neural tube defects with a mutation in the Hectd1 gene. By comparing the cell movements in this model with those that occur during normal development, we aim to elucidate how disruption of cranial mesenchyme expansion can result in failure of neural tube closure.
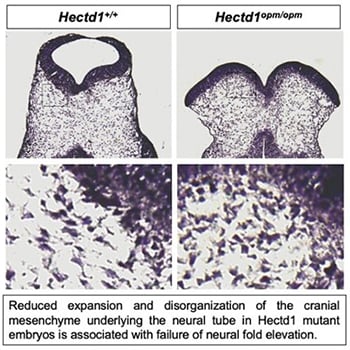
To study cranial mesenchyme morphogenesis we utilize combinations of innovative tools, including:
- Advanced imaging approaches to visualize, at unprecedented resolution, expansion of the cranial mesenchyme in real time during neural fold elevation
- Fluorescently labeled probes to elucidate spatial and temporal patterns of pathogenic protein production
- An ex vivo cranial mesenchyme explant assay amenable to experimental manipulation
- A novel allelic series of preclinical model lines
- Pharmacological inhibitors to test the role of molecules implicated in failure of cranial mesenchyme expansion and neural fold elevation
These innovative tools are combined with well-established, conditional genetic, histological and embryological approaches to evaluate expansion of cranial mesenchyme during normal neural fold elevation and reveal how this process goes awry during failed cranial mesenchyme expansion responsible for neural tube defects. This information is then used to determine the impact of predicted pathogenic sequence variants identified in human neural tube defects cases and ascertain if variants disrupt gene function and contribute to abnormal development in these patients.
How do changes in maternal dietary intake of Vitamin A interact with genetic mutations to cause congenital heart defects?
Understanding how cardiac progenitor cell populations are misspecified in congenital heart defects is of major importance for elucidating the origin of these malformations as well as informing stem cell work. Outflow tract defects account for 30% of all congenital heart defects and aortic arch malformations are reported in 1% to 2% of the population. Distinct second heart field-derived progenitor populations contribute to the outflow tract versus the aortic arch, yet our ability to distinguish these progenitors in higher vertebrates, our knowledge of how different progenitors within the second heart field are specified and how altered specification contributes to divergent CHDs remains fragmented. Retinoic acid, the biologically active metabolite of vitamin A, is essential for establishment of the posterior boundary of the second heart field and plays a poorly defined role in partitioning the second heart field into different progenitor domains. Importantly, maternal vitamin A deficiency or teratogenesis are linked to increased risk for congenital heart defects affecting the outflow tract and aortic arch.
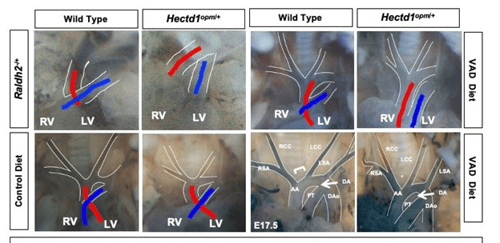
The Zohn Laboratory is addressing these gaps in knowledge utilizing novel models of congenital heart defects that interact with vitamin A signaling. Our data indicate that altering maternal vitamin A intake can preferentially impact the outflow tract or aortic arch depending upon whether signaling is increased or decreased. We hypothesize that this differential activity is due to alterations in specification of outflow tract and aortic arch progenitors in the second heart field. These studies have the potential to elucidate how environmental factors alter progenitor cell specification resulting in congenital heart defects.
What are the mechanisms underlying variability of congenital heart defects in genetic syndromes?
The developing embryo has the remarkable ability to buffer insults allowing for normal development in the face of genetic and environmental influences; however, when insults exceed a disease threshold, birth defects can result. Our goal is to better understand how these buffering systems mediate the interplay between genes and environment leading to structural birth defects and if disruption of these buffering systems contributes to birth defects in genetic syndromes. Resistance to perturbations is known as developmental robustness and miRNA-mediated negative feedback loops potentially play key roles in buffering genetic and environmental insults. Disruption of miRNAs results in increased susceptibility to environmental factors in model organisms but the role in birth defects in humans and preclinical models of human birth defects remains unknown.
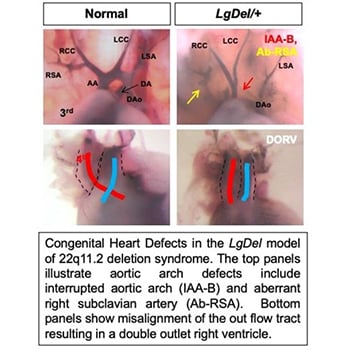
The Zohn Laboratory is investigating whether defective miRNA-mediated buffering systems might drive phenotypic variability observed in genetic syndromes. Our data establish that dietary supplementation of the maternal diet with vitamin A (within recommended levels for pregnancy) can modify heart development. In light of the role of miRNAs in tuning developmental pathways, we are testing whether reduced miRNA processing can create a bottleneck in miRNA processing, impacting vitamin A buffering, sensitizing the embryo to genetic and environmental perturbations. We postulate that reduced buffering capacity transforms nontoxic vitamin A exposures into teratogenic doses through failure to rapidly engage miRNA-mediated negative feedback mechanisms to modulate signaling. These experiments will provide a new understanding of the mechanisms mediating gene-environment interactions, insight that can be directly translated to prevention of birth defects.