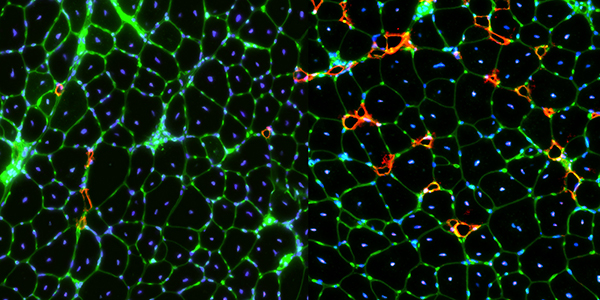
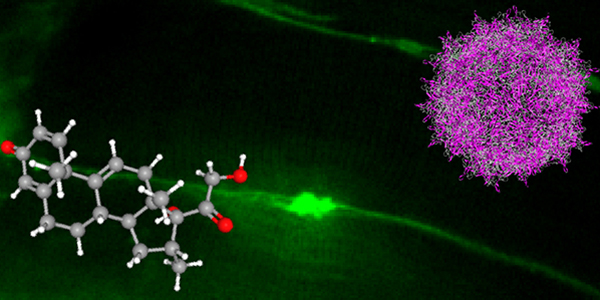
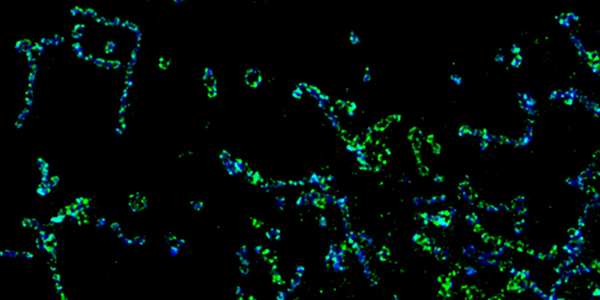
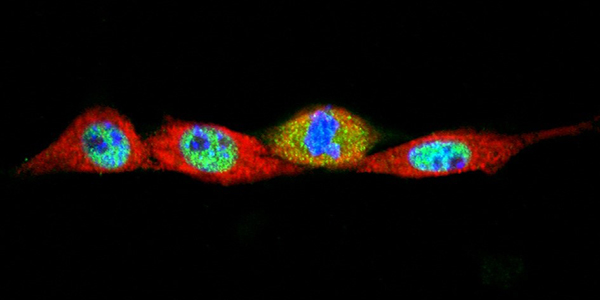
Our studies are uncovering the role of several cellular compartments, calcium-binding proteins and lipids in cell repair. These include the role of lysosomes, which are the major calcium-triggered secretory compartments in mammalian cells. Defect in fusion of lysosomes impairs release of the lipid-modifying lysosomal enzyme, acid sphingomyelinase (ASM), and results in diseases such as dysferlinopathy or limb-girdle muscular dystrophy 2B (LGMD2B). Fusion of lysosomes and other calcium-regulated organelles in cells is controlled by multiple calcium-binding proteins, including annexins, dysferlin, and synaptotagmins, that we have examined for their role in calcium-triggered vesicle fusion during cell repair. Our work has also identified the need for the cytosol and endoplasmic reticulum to buffer injury-triggered calcium increase to control cell repair. A deficit in this process contributes to limb-girdle muscular dystrophy 2B (LGMD2L). Additionally, we find that beyond triggering membrane fusion, calcium also triggers membrane scission by the endosomal sorting complex required for transport (ESCRT) machinery, which is required for cell repair. This is regulated by the calcium-brining protein Apoptosis linked gene-2 (ALG-2) to direct scission and shedding of the damaged parts of the plasma membrane to help close a wound. We are continuing to study additional regulators of calcium-dependent membrane trafficking and their role in cell repair.
Further reading:
- Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion
- Dysregulated calcium homeostasis prevents plasma membrane repair in Anoctamin 5/TMEM16E-deficient patient muscle cells
- Injured astrocytes are repaired by Synaptotagmin XI-regulated lysosome exocytosis
- Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair
- Early Endosomes Undergo Calcium-Triggered Exocytosis and Enable Repair of Diffuse and Focal Plasma Membrane Injury